用户名
UID
Email
密码
记住
立即注册
找回密码
只需一步,快速开始
微信扫一扫,快速登录
开启辅助访问
收藏本站
快捷导航
门户
Portal
社区
BBS
资讯
会议
市场
产品
问答
数据
专题
帮助
签到
每日签到
企业联盟
人才基地
独立实验室
产业园区
投资机构
检验科
招标动态
供给发布
同行交流
悬赏任务
共享资源
VIP资源
百科词条
互动话题
导读
动态
广播
淘贴
法规政策
市场营销
创业投资
会议信息
企业新闻
新品介绍
体系交流
注册交流
临床交流
同行交流
技术杂谈
检验杂谈
今日桔说
共享资源
VIP专区
企业联盟
投资机构
产业园区
业务合作
投稿通道
升级会员
联系我们
搜索
搜索
本版
文章
帖子
用户
小桔灯网
»
社区
›
C、IVD技术区
›
生物信息服务
›
发生物信息学文章,只做生信行不行?为啥都做实验呢? ...
图文播报
2026庆【网站十三周
2025庆【网站十二周
2024庆中秋、迎国庆
2024庆【网站十一周
2023庆【网站十周年
2022庆【网站九周年
返回列表
查看:
7364
|
回复:
5
[分享]
发生物信息学文章,只做生信行不行?为啥都做实验呢?
[复制链接]
空白派
空白派
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
雷达卡
发表于 2025-5-10 11:24
|
显示全部楼层
|
阅读模式
登陆有奖并可浏览互动!
您需要
登录
才可以下载或查看,没有账号?
立即注册
×
发生物信息学文章,只做生信行不行?为啥都做实验呢?
原文地址:https://www.zhihu.com/question/543423420
回复
使用道具
举报
提升卡
检验医师
检验医师
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
雷达卡
发表于 2025-5-10 11:25
|
显示全部楼层
DNA起始量低,为何不能尽可能多的增加PCR循环数?
为什么要严格控制DNA建库中PCR循环数?
众所周知,
DNA建库过程中PCR循环数是决定测序文库质量好坏的重要因素之一
。尤其是DNA起始量较低时,如果
循环数过少
会导致
文库产出不足
,
无法满足测序要求
;而如果
循环数过多
又会导致
过度扩增
、
偏好性增加
、
测序duplicate增加
、
嵌合产物增加
、
扩增突变积累
等诸多不良后果。因此,
严格控制PCR循环数对于DNA建库过程尤为重要
。
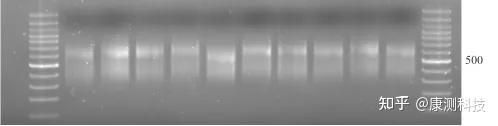
什么是嵌合产物?
PCR反应后期,引物通常会先于dNTP被耗尽。此时,过多的循环会造成扩增产物解链后进行非特异性退火,产生非互补链交叉的退火产物 ,这便是嵌合产物。这些产物在依赖电泳的检测方式中迁移速率比较慢,在高分子量区域呈现弥散分布。
过度扩增(存在嵌合产物)案例
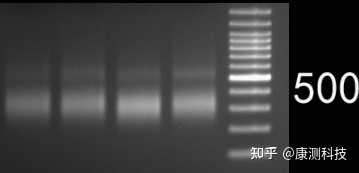
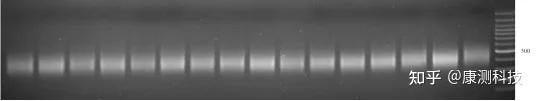
正常扩增案例
P.S. 康测科技二代测序文库正常片段分布主要在300-500 bp,而过度扩增的文库500 bp以上出现了一个嵌合产物的特异性条带
PCR循环数过多带来的嵌合产物对测序有何影响?
嵌合产物主要会影响DNA文库定量方法的选择
。假设使用吸光度方法的Nanodrop仪器测得的文库浓度为20 ng/μL;当使用基于识别双链DNA的荧光染料的Qubit仪器来进行文库定量时,定量结果可能只有5 ng/μL,远远低于实际值。所以对于有嵌合产物的文库,只有使用qPCR方法对文库定量才能规避这种问题,因为在qPCR定量过程中包含变性过程,会解开嵌合产物,仍然可以准确定量这种过度扩增的文库。
而如果不用qPCR定量,使用Qubit定量结果会产生什么后果?假设以5 ng/μL进行文库测序pooling时,会以远低于正常准确浓度进行投入,造成过多文库DNA混入,由于嵌合产物在变性后能够与Flow Cell正常结合并被测序,
虽然对测序过程无显著影响,但过多单一文库会影响pooling池子的平衡,大大压缩其他文库的数据量产出
。
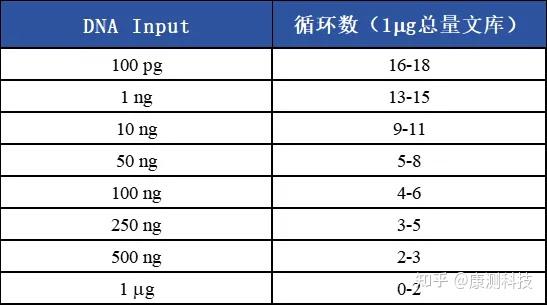
我司项目经验
下表列举了当使用不同DNA起始量,获得1 μg总量文库推荐的扩增循环数;
我司始终坚持严格控制建库过程中PCR循环数,避免嵌合产物的产生,并保障客户测序数据的正常产出。
回复
支持
反对
使用道具
举报
大力水手
大力水手
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
雷达卡
发表于 2025-5-10 11:25
|
显示全部楼层
当然可以啊,不做实验也能发生信,关键区别在于如何发高分。
仔细研究了2分左右的生信文章,发现它们主要聚焦在差异基因的筛选、功能富集分析、基因表达水平的检验、预后模型的建立和评估等等。
这些都是常规的生信分析思路,虽然对你的动手能力没啥要求,但因为创新程度很低,收益也就不会高。
说到底还是套路重复,方法不及时迭代,再加上大环境越来越卷,现在纯生信想发高分可以说是越来越难,不补点实验很难被大杂志认可。
动手能力不强,还想要搞搞科研,发SCI,必须升级方法和套路!
下面我就给大家介绍3个2022年不做实验发SCI的蓝海领域。并且给大家送上免费的教程资源合集,需要的直接划到末尾。
1
影像组学
如果只能三选一,那么影像组学绝对是今年必须抢占的发文先机。
影像组学的概念自诞生以来就在不断完善,一个比较成熟的定义是,影像组学指从CT、PET或MRI等医学影像图像中高通量地提取并分析大量高级且定量的影像学特征。
通俗地来讲,我们可以把它拆成“影像”和"组学”两个部分进行理解。
所谓“影像”,通常指放射影像,主要包括了CT、MR影像等;所谓“组学”(Omics),其实就是把与研究目标相关的所有因素综合在一起作为一个“系统”来研究。
影像组学本质上来说其实是一种
分析思路方法
,从临床问题出发,最后回到解决的临床问题。也正因此,在发文的时候,
影像组学对实验并没有硬性要求
。
所以,你会看到——
虽然不做实验,但是入门级别的影像组学都能发6分;
虽然不做实验,但是影像组学+单细胞多组学能轻松上10分;
虽然不做实验,但是生信分析加点影像组学就能发到17+……
影像组学自2016年以来一直是热点,因此发文量飙升,但规范的影像组学论文依然能发到非常好的杂志,这是因为影像组学的价值越来越被临床认可。
随着数据越来越标准,研究越来越规范,
影像组学,或者影像组+人工智能在肿瘤学等领域会愈发大放异彩。
如果你恰好还会一点
机器学习
,那在发SCI的路上可以说是神挡杀神,佛挡杀佛了。
2
空间转录组
想做单细胞测序,就不要放过空间转录组。
简单来讲,空间转录组是既能够保留组织空间位置信息又能够获得不同组织空间位点转录信息的一种技术,以10x Genomics Visium技术为例,它融合了组织病理学、基因芯片、高通量测序、生物成像技术。
空间转录组测序技术目前蓬勃发展,在现有的高通量检测技术领域,它已为科学研究的发展提供了前所未有的技术支撑。
那么,为什么要把空间转录组和单细胞结合?
我们都知道单细胞测序是大腿。但是它其缺憾,那就是无法检测到空间信息,因为单细胞测序技术在将实体组织解离成单个细胞的过程中,
不可避免地丢失了空间信息
。空间转录组则可以
提供空间位置信息
,完美补偿这一点。
但是目前来看,
空间转录组并没有做到完全的单细胞
,其每一个spot大概会有1-10个细胞左右,所以并不是完全的“单细胞”。
因此,当我们通过算法把单细胞的精度和空间转录组联合运用起来时,我们就拥有了一个双剑合璧的完美上分利器!
这俩加起来有多牛呢?
可以说,空间转录组绝对属于新的测序技术的新宠,2021年有
160篇
文章发表!
其中,Cell、Nature、Science也不乏空间转录组+单细胞的身影,
54+Nature子刊,40+Cell,13+可复制生信高分模板……
如果说单细胞的福利是在前两年,那么当下的福利就理所应到的在空间转录组身上。buff叠起来,文章发起来!你要是能染几张片子简单验证一下,那大杂志就更是认可有加了!
3
机器学习
仅仅2021年一年,生信+机器学习的套路已经在10+SCI上发了近60篇文章!
所谓机器学习,是指利用算法来检测数据中的模式,而不需要明确的指示。一个学习系统可以利用训练数据集,学会找出输入信息(例如图片)的特征与输出信息(如标签)之间的关联。
机器学习在生信分析中的运用越来越常见,越来越重要。
比如在
基因组数据
方面,用深度学习检测突变逐渐成为主流方法。
比如
群体基因组学领域的PRS
,用于基于基因组数据预测生物性状,这就是个典型的机器学习预测问题。
比如
非肿瘤研究
中,可以用机器学习来评估我们诊断标志物的诊断效力。
临床预测模型、筛基因、二代测序、代谢通路、非编码RNA分析、蛋白质结构功能预测、疾病亚型分型、术后预测……机器学习在这些方面的运用已经越来越多见了。
我为大家整理了151页免费的机器学习攻略合集,从什么是机器学习,到超多实用算法详解,再到教你如何将机器学习与生信结合,手把手带领大家走上发生信高分的康庄大道。同时附赠0基础医学科研小白SCI发文训练营免费名额,点击免费领取。
超多实用算法一次get
KNN算法、Kmeans算法、决策树算法、贝叶斯公式、线性回归、非线性模型、神经网络、mlr3包……
12篇教程文详解如何用机器学习发高分
在机器学习的帮助下,如何结合生存信息对基因表达情况进行变量的筛选与降维处理?非肿瘤研究中,如何用机器学习来评估诊断标志物的诊断效力?如何用机器学习改进临床预测模型……
8天领悟5分SCI训练营
回复
支持
反对
使用道具
举报
大力水手
大力水手
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
雷达卡
发表于 2025-5-10 11:26
|
显示全部楼层
当然可以啊,生信又不止数据挖掘。
你可以针对一个命题去做深度学习,然后去做预测,基于你的模型然后讲故事,也可以发很好的文章啊,像是这样:
Deep learning modeling m 6 A deposition reveals the importance of downstream cis-element sequences - PubMed (nih.gov)
你也可以针对一个pipeline用到的各种各样的工具,去做比较学习,这样的文章也很受欢迎啊,像是这样:
RiboReport - benchmarking tools for ribosome profiling-based identification of open reading frames in bacteria - PubMed (nih.gov)
你能够整合一些已有的工具,做整合的多组学的分析,什么基因组转录组,蛋白组全部往里面扔,再加点什么机器学习什么的,就可以发很好的文章啊,像这种:
Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer - PubMed (nih.gov)
现在单细胞不是很火吗,感觉随便基于单细胞的分析做点能够方便别人分析的R包,python包什么的,感觉也可以发很好的文章啊,我不是做这方面举不出例子来。
我不知道上面都在讨论些什么,只做生信当然可以发出很好的文章来。现在只做数据挖掘确实不好发了,但生信又不是只有数据挖掘,我觉得数据挖掘只是生信工作里面最基础的工作啊,生信可以做的方向还有很多,而且目前也都很热门啊。
更新:近期最让我敬佩的一篇,一个共计17.2 KB 的R包发了
Nat Methods
不服不行,
A computational framework for the inference of protein complex remodeling from whole-proteome measurements - PubMed (nih.gov)
生信想发好文章,要么你很懂生物,要么你很懂数学,你啥也不会就知道公共数据库找几个差异基因想发篇好文章想屁吃呢。
继续更新:基于UKbiobank数据的基因组分析:
Large-scale exome sequencing identified 18 novel genes for neuroticism in 394,005 UK-based individuals
孟德尔随机化,虽然已经控过一次水的,但是高质量的还是能发的:
Mammographic density mediates the protective effect of early-life body size on breast cancer risk
。
药靶的文章更多一些,这个领域确实有点无聊了:
Identification of novel therapeutic targets for chronic kidney disease and kidney function by integrating multi-omics proteome with transcriptome
,
Proteogenomic Data Integration Reveals CXCL10 as a Potentially Downstream Causal Mediator for IL-6 Signaling on Atherosclerosis
算法框架,我数学太差了没看具体做了什么:
Discovery of sparse, reliable omic biomarkers with Stabl
回复
支持
反对
使用道具
举报
继续前进
继续前进
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
雷达卡
发表于 2025-5-10 11:26
|
显示全部楼层
可以,但是可信度会存疑。纯生信文章现在并不吃香,想要提升影响因子肯定辅以实验验证。很多CNS正刊、子刊的生信文章都会最后落在是实验来验证或者拓展内容。
其主要原因是论证观点不仅需要多方面佐证你的观点,同时还需要从不同的角度来印证你的结论或者方法的有效性和可信度,这样才会更有效的证实你的conclusion。
同时,生信虽是交叉学科,但毕竟还是“生”字当头,其本身发展就建立于实验基础之上,很多理论知识和公知都是建立在实验科学的发展。并且,“seeing is believing”仍然固化于心(笔者持怀疑态度),所以很多reviewer还是希望你的论证都能以实验佐证。
所以,添加一些实验说不定能成为你文章的点睛之笔呢(也说不定reviewer想叫你加,不加都得加)。
回复
支持
反对
使用道具
举报
同花顺
同花顺
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
雷达卡
发表于 2025-5-10 11:27
|
显示全部楼层
可以只做生信,比如开发新的方法,或是提供新鲜的分析思路(旧数据玩出新花样)。
“都做实验”是因为现在很多生信分析流程已经十分成熟,教程也越来越多,门槛逐渐降低,自然发好文章的要求也水涨船高;此外,分析的结果只有经过检验才更有说服力,很多高水平期刊都会要求实验证据。
生物信息学是分析数据(主要是组学数据)不假,但数据本身也需要做一些实验才能获取。
如果只想用公共数据来做,在没有实验支撑的情况下,想发好的文章(获得好的结果),对个人的学术水平和技术水平的要求都非常非常高。
最后,我贴两篇最近发表在Nature Genetics上的使用公共数据进行分析的生信文章(其实应该说是两篇方法开发类的文章),供你参考。
1、主要用的是来自TCGA的406个ATAC-Seq癌症样本
https://doi.org/10.1038/s41588-022-01075-2
该文章提出了新的关联分析方法RWAS(对标GWAS、TWAS理解即可)
2、主要用到的数据包括:
4DN计划的4DNFI9GMP2J8、4DNFI643OYP9、4DNFILP99QJS;NCBI下载的GSE137372、GSE66383、GSE78109 ;EBI下载的PRJEB5236.
https://doi.org/10.1038/s41588-022-01065-4
该文章提出了一个深度学习模型Orca,可以在k碱基直至染色体尺度预测基因组三维结构。
回复
支持
反对
使用道具
举报
返回列表
发表回复
高级模式
B
Color
Image
Link
Quote
Code
Smilies
您需要登录后才可以回帖
登录
|
立即注册
本版积分规则
发表回复
回帖后跳转到最后一页
关闭
官方推荐
/3
AI助手<小桔子>来了!
欢迎来交流,可以回答IVD行业各类问题!
查看 »
IVD业界薪资调查(月薪/税前)
长期活动,投票后可见结果!看看咱们这个行业个人的前景如何。请热爱行业的桔友们积极参与!
查看 »
小桔灯网视频号开通了!
扫描二维码,关注视频号!
查看 »
返回顶部
快速回复
返回列表
客服中心
搜索
洽谈合作
关注微信
微信扫一扫关注本站公众号
个人中心
个人中心
登录或注册
业务合作
-
投稿通道
-
友链申请
-
手机版
-
联系我们
-
免责声明
-
返回首页
Copyright © 2008-2024
小桔灯网
(https://www.iivd.net) 版权所有 All Rights Reserved.
免责声明: 本网不承担任何由内容提供商提供的信息所引起的争议和法律责任。
Powered by
Discuz!
X3.5 技术支持:
宇翼科技
浙ICP备18026348号-2
浙公网安备33010802005999号
快速回复
返回顶部
返回列表

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-5-10 11:24
发表于 2025-5-10 11:24


 提升卡
提升卡